
On May 14, 2025, the Food and Drug Administration approved belzutifan (Welireg, Merck & Co., Inc.) for adult and pediatric patients 12 years and older with locally advanced, unresectable, or metastatic pheochromocytoma or paraganglioma (PPGL).
Our Mission
Resources
Our mission at the SDHB PheoPara Coalition is to provide resources to both patients and healthcare providers. Through our website and organized events, we provide information on these devastating diseases and what specifically is being done that could benefit patients both now and in the future. We provide information on the disease itself, treatment centers and can often put a patient in touch with a qualified and experienced physician or center in their area.
Research / Grants
The SDHB PheoPara Coalition is active in sponsoring and supporting research in this underserved area with the hope of finding better treatment options and/or a cure. Grant applications are carefully vetted to ensure that the research and projected outcomes will have a benefit on the SDHB germline mutation and its specific impact on patients. Typically, we have in the past or are currently supporting two or three major studies in this regard.
Awareness
Pheochromocytoma’s and Paraganglioma’s are indeed rare tumors when compared to other well known cancers and so it is very important that we continue to shine the spotlight on these devastating diseases. We use various communication vehicles to achieve this goal which include but is not limited to the use of our website, annual gala and fund raising event, sponsoring of educational events and last but not least social media, i.e. Facebook, Instagram, LinkedIn, etc.
The SDHB PheoPara Coalition is made possible through charitable contributions by you. Please donate today.
What's In The News...

FDA
May 14, 2025 | No Comments
Categories :
FDA approves Belzutifan for Pheochromocytoma or Paraganglioma
Events and Annual Gala
View event details, register and purchase tickets for upcoming annual gala. (Click on the tiles below for more information)




Current Projects and Latest Update
The SDHB PheoPara Coalition is active in sponsoring and supporting research in this underserved area with the hope of finding better treatment options and/or a cure. (Click below for latest updates. Please use arrows to see more institutions)
University of Auckland – Lead Investigator Daniel Conole
SDHB Pheo Para Coalition Research Project update: To establish a cell-based model for the discovery of new molecules that inhibit the degradation of altered SDHB and rescue its tumor suppressor function in PPGL
Summary:
In our SDHB PheoPara Coalition funded work, we aim to examine whether we can chemically stabilize pathogenic Succinate Dehydrogenase subunit B (SDHB), a tumor suppressor gene. Pheochromocytomas and Paragangliomas (PPGL) patients harboring a pathogenic mutation in the SDHB experience increased cancer metastasis and limited treatment options. This is because SDHB pathogenic variants are rapidly degraded in PPGL, which inhibits its ability to carry out is usual tumor suppressor function.
With the generous support from the SDHB Pheo Para Coalition in 2024, we established two human succinate dehydrogenase (SDHB) deficient cell models in our lab. With additional support from the US Department of Defense, we are now using genetic engineering approaches, such as inducible expression models, to examine what functional effects of artificially stabilizing SDHB in our models. Our preliminary data suggests that while wildtype SDHB activity can be rescued by protein stabilization, the effectiveness of this approach on altered forms of SDHB found in patients will be dependent on the type of alteration. This is an ongoing area of research, and we are actively exploring different SDHB deficient models and different clinically relevant SDHB mutants.
In a separate but related project, funded by New Zealand’s Maruice Wilkins Centre for Molecular Biodiscovery, we are now using these SDHB inducible expression models and functional genomics (e.g. pooled CRISPR screening) to discover what proteins within the cell are responsible for either enhancing or degrading altered SDHB. Identification of proteins these will direct our drug discovery efforts.
With continued support from the SDHB coalition in 2025, we are exploring new bifunctional therapies for the treatment of SDHB-deficient cancers. Lutetium-177-Dotatate peptide receptor radionuclide therapy (LuTATE PRRT) is a clinically approved and selective bifunctional treatment for PPGL patients (including those that are SDHB-deficient), however there are limitations with its use. Using publicly available proteomics and in-house transcriptomics data, we are designing, synthesizing, and evaluating the selective cancer cell killing potential of new bifunctional compounds called Regulated Induced Proximity targeting chimeras (RIPTACs). RIPTACs are heterobifunctional small molecules that elicit a stable ternary complex between a protein over-expressed in tumor cells and a protein essential for cell survival. This leads to “hold-and-kill” effect, causing selective accumulation of the RIPTAC in cancer cells and selective cell killing versus healthy cells.
We thank the SDHB coalition and their generous donors for their support thus far and look forward to continuing our work together.
Read More
Linkoping University – Lead Investigator Oliver Gimm
Identification of new drugs for the treatment of malignant pheochromocytomas and paragangliomas (PPGLs)
This project is focused on the identification of potential therapeutic drugs for the treatment of malignant Pheochromocytoma and Paraganglioma (PPGLs). The project is structured around two primary aims:
Aim #1: to use the human pheochromocytoma cell line (hPheo1) that will be genetically modified (by introducing mutations associated with malignancy, i.e., SDHB) to screen libraries of drugs already approved for other indications regarding their effect on hPheo1 (drug repurposing),
Aim #2: to analyze whether the somatic mutations in human pheochromocytomas can be identified in liquid biopsies (e.g., cell-free DNA). This would help regarding the choice of the drug identified under Aim #1 to be the most effective one for individual patients diagnosed with malignant PPGL.
Progress regarding aim #1: The project has initiated a collaboration with the Chemical Biology Consortium Sweden (CBCS) at SciLifeLab, focusing initially on the SDHB gene. After the successful knockdown of the SDHB gene in the hPheo1 cell line using CRISPR-cas9, the mutated cell line has been characterized using various techniques such as DNA sequencing, DDPCR, western blot, and microarray gene expression analysis.
The current goal is to identify approved drugs that specifically target mutated pheochromocytoma cells without affecting wild-type cells. This involves screening a collection of about 8,000 clinically active compounds, including approximately 1,200 FDA-approved drugs.
The project is conducting extensive dose-response experiments using both 3D and 2D cell cultures to validate potential hits. Cell viability and microscopic imaging are being used as assays to assess the effectiveness of these compounds.
The project commenced with the transfer of assays to prescreening, a crucial step for establishing the hit threshold. Subsequently, the primary screening experiment was conducted, testing the entire set of compounds at a concentration of 10µM. Based on the previously set hit threshold, 704 compounds were selected.
The project then advanced to hit validation, which was performed at three different concentrations of the selected compounds. Compounds were deselected if they exhibited less than 40% cell inhibition and showed the same or higher effect in wild-type (WT) cells. From this process, 22 compounds were chosen as candidates for dose-response curves. The dose-response results revealed that six compound batches and five individual structures demonstrated a significant difference in cytotoxicity between the two cell lines, with higher levels of cell killing observed in the knockdown (KD) cells compared to the WT cells. The remaining compounds either showed similar levels of cell killing in both lines or greater activity in the WT cells.
Based on these findings, potential candidates for further follow-up experiments were selected, considering both functional annotations and structural analogues.
Following this, the project will proceed with testing the analogues at various concentrations. Subsequently, hit validation of the resulting compounds from the screen and analogue testing will be performed. This includes gene expression analysis, Western blotting, and in vivo studies.
Progress regarding aim #2: In a concurrent line of investigation, we pursued our secondary objective, which involved the detection of somatic mutations in PPGLs in order to facilitate mutation‐specific drug treatment. Circulating free DNA (cfDNA) extracted from patients with PPGLs is anticipated to mirror the mutation status of the susceptibility genes. While examining frozen blood samples from pheochromocytoma tumors, we were able to successfully pinpoint different somatic mutations in cfDNA. Our approach involved the use of two sets of mutation-specific primers and Sanger sequencing.
Read More
University of Arizona – Lead Investigator Dr. James Bibb
NOVEL MECHANISMS and THERAPEUTICS FOR SDHB MUTATION-DRIVEN PHEOCHROMOCYTOMA
Genetically determined metabolic dysfunctions or inborn errors of metabolism disrupt normal resting or senescent state bioenergetic sensing, causing cells to undergo uncontrolled proliferation. Such genetic disorders are typified by loss-of-function mutations in succinate dehydrogenase (SDH), which both impairs metabolism and causes uncontrollable cell proliferation in adrenal and paraganglia glands resulting in two related recalcitrant, debilitating, and lethal cancers, pheochromocytoma (PC) and paraganglioma (PG).
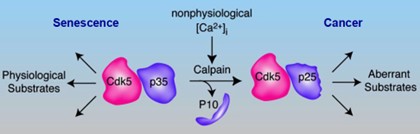
How these mutations cause these cancers has been poorly understood. Thanks to SDHB PheoPara Coalition grant support, in 2022 we reported a new pathway by which deficiencies in SDHB mutations that cause buildup of the metabolic intermediate succinate, hyperactivates succinate receptors, resulting in loss of adrenal chromaffin cell calcium homeostasis. This activates the protease, calpain, to cleave the protein p35 which serves as the activating cofactor of a protein kinase named Cdk5. Conversion of Cdk5/p35 to Cdk5/p25 renders the kinase aberrantly active and has the downstream effect of inactivating a critical bioenergetic sensor, AMP-kinase. This releases cells from the resting state and they uncontrollably divide and multiply. Thus, we discovered an important mechanistic signaling cascade by which SDHB mutations give rise to PC. We also introduced the first inducible/arrestable clinically accurate animal model of PC, and demonstrated a newly discovered anti-Cdk5 drug, MRT3-007 which halted or regressed PC tumors across several preclinical models.
These advances underline the importance of continuing this research in two critical directions. First, it is now clear that Cdk5 is a principal drug discovery target for PC treatment. Second, better mechanistic understanding of how aberrant Cdk5 causes SDHB mutation-linked PC/PG will reveal additional specific targets for therapy development. With SDHB PheoPara Coalition support, we are aggressively pursuing these two goals. We aim to bring biomarker guided patient specific precision medicine cures for PC and PG. We are aggressively leveraging the innovative systems and tools we have developed to better understand the disease mechanisms and uncover new targets for treatment.
Like all kinases, Cdk5 catalyzes protein phosphorylation, transferring a phosphate from ATP to specific sites on proteins. Our current anti-Cdk5 drugs act at the enzyme’s ATP binding site and show efficacy in animal models of PC, as proof of principle. However, they are not adequately specific and exhibit toxicity at doses 4-fold higher than those found to be minimally effective. To achieve a wider treatment dose window, new drugs with different modes of action are needed. We are now screening a novel peptoid inspired, conformationally constrained oligomer (PICCOs) bead display compound library for compounds that will block Cdk5/p25 activity through allosteric interactions beyond the ATP binding site or through protein-protein interaction disruption. Preliminary studies indicate that this approach is producing hits for compounds that bind tightly to Cdk5/p25. We are refining the screen and hope to have our first validated compounds to begin preclinical testing soon. Effective and safe compounds will be paired with a diagnostic biomarker assay designed to identify Cdk5-driven tumors and thus direct these therapies as a novel precision medicine approach.
Concomitantly, we are aggressively pursuing pathways that are both upstream and downstream of Cdk5/p25 so that new targets may be identified and adapted to drug library screening. Blocking p35 conversion to p25 may be an effective treatment strategy. Thus, we are creating a new system to identify the proteins that interact with p35 (i.e., the p35 tumorigenic ‘interactome’) when succinate receptors are activated. We hypothesize that disruption of key binding partners will prevent efficient calpain cleavage of p35. We also discovered novel pathways by which aberrant Cdk5 activates cancer causing gene expression. These gene regulatory mechanisms are critical for neoplastic programming downstream of Cdk5/p25. Studying these will yield new and specific therapeutic targets. We believe our research will lead to cures for PC and PG. However, this work is challenging, requires resources, and would not be possible without SDHB PheoPara Coalition support. Thank you for your support.
Read More

University of Melbourne – Lead Investigator Dr. Richard Tothill
A5 SDHB Genomics Study
The A5 SDHB Genomics study is an ambitious multi-national research project involving comprehensive analysis of metastatic SDHB-associated pheochromocytoma and paraganglioma (PCPG).
The study has these key objectives:
- To elucidate the biology of SDHB PC/PGL by characterizing recurrently altered genes, gene pathways and cell types in tumors.
- To identify either single biomarkers or biomarker gene sets that can predict the likelihood of metastatic progression.
- To identify new therapeutic targets representing single genes, proteins or signalling pathways.
The team is led by A/Prof Richard Tothill at the University of Melbourne, and patient samples and clinical data have been accrued from 12 institutions across six countries. A/Prof Tothill’s team has applied genome-wide multi-omics analysis to more than 100 PCPG tumors including whole-genome DNA sequencing, DNA methylation profiling and cutting-edge single cell analysis. The work has been generously funded by the National Health and Medical Research Council (Australia), Pheo-Para Coalition (USA), Pheo-Para Alliance (USA) and Paradifference (Sweden).
A recent study published by the team in the journal Nature Communications described analysis of PCPG tumors using single-cell genomics. The method enabled simultaneous gene-expression analysis in thousands of individual cells from patient tumor samples. The study involved analysis of 30 PCPG tumors representing diverse genetic backgrounds (including SDHB carriers) as well as two normal adrenal tissues totalling more than 109,000 cells.
Key findings from the single-cell genomics study included an elevation of normal cell types associated with blood vessel formation in tumors, which is linked to aberrant hypoxia pathway signalling with some unexpected observations made in PCPG carrying MAML3-fusions, that also have some of these features. Other cell types of interest included immune cells such as macrophages that were abundant in some tumors. A support cell type called a sustentactular cell, commonly seen in PCPG tumors and normal adrenal tissues, were also characterised in great detail. A comparison of cancerous cells found in tumors to normal healthy adrenal chromaffin cells showed associations between tumor genotype and developmental stages in the adrenal gland but that tumor cells also have very unique transcriptional changes based on underlying mutated genes. The team also combined their single-cell analysis with data from 735 tumors profiled using “bulk tissue” analysis to conduct the largest single analysis of its kind to date. The data was used to refine molecular subtypes of PCPG and find gene-expression changes associated with metastatic progression in SDHB-associated PCPG. Finally, promising new diagnostic and therapeutic targets were identified, including the G-protein couple receptor GPR139, that had elevated expression in pseudohypoxic PCPG including metastatic SDHB-associated PCPG.
Further analysis of the other data types generated during the A5 SDHB Genomics Study is still underway and currently being finalised for preparation of manuscripts. The data will be valuable resource made available to other researchers once published. Indeed, the single cell genomics data has already been made publicly available to other research groups for their own analysis.
Read More
The Broad Institute – Project Lead, Bill Sellers
Rare Cancer Dependency Map Initiative
Thanks to the generous support of our partner organizations, three years ago the Broad Institute of MIT and Harvard launched an initiative to address key knowledge gaps and accelerate therapeutic discovery across rare cancers. The ultimate goals of this effort, the Rare Cancer Dependency Map Initiative, are to (1) leverage our established fresh tissue acquisition pipeline for any patient with a rare cancer to donate living tissue for the derivation of patient models, and
(2) create a leading rare cancer translational research platform focused on systematically identifying drug repurposing hypotheses and genetic dependencies of rare tumors. At the end of these 3 years, we feel we have made significant progress among the consortium as a whole.
After three years of this Initiative, we’d like to express our gratitude for our partners’ support. The following 3 foundations were involved in providing research support for our Rare Cancer Dependency Map Initiative:
- ParaDifference Foundation
- PheoPara Alliance
- SDHB PheoPara Coalition
Pheochromocytoma and Paraganglioma Update – July 2022
Though we did not generate any long-term cancer cell models for pheo/para diseases, our ultimate goal was to apply the past 8 years of the CCLF team’s model derivation experience and determine the feasibility of growing cancer cell models for pheo/para (PCPG) diseases. To achieve this, we focused on onboarding and identifying high quality viable tumor samples as well as systematically iterating model derivation strategies.
Summary & Conclusion
Listed below is a summary of all milestones to date:
- We have established a tissue acquisition pipeline to rapidly acquire patient samples from the direct-to-patient mechanism through RCRF and have optimized the sample acquisition workflow with Dr. Arthur Tischler. Additionally, all 37 received samples have been fully genomic QC’ed to understand the sample quality and genomic driver events.
- After 3 iterations of rich-media designs in over 153 different conditions, we observed that several growing cultures can be propagated beyond passage 5. We are currently testing different strategies (2D/3D/coating/media boosting) to improve the likelihood of generating long-term cell models that can be shared with the research community in the near future.
- Using the CCLF cancer detection sequencing panel (400 cancer genes and copy numbers), two genomically verified PCPG cell models (currently at passage 10) are currently being expanded to potential long-term models.
We understand this has been a very challenging mission as there are no PCPG cancer cell lines in the world yet; therefore, we worked closely with Dr. Tischler’s group and iterated our strategies from other successful model generation attempts. More work is needed to be done, including optimizing the cell model propagation ability in reducing slow doubling time, in order to have a further understanding of the characterization of cell model fidelity, etc. We thank all three foundations for your wonderful partnership in these efforts for the past 3 years and the entire pheochromocytoma and paraganglioma community for their support of this project.
Read More
University of Florida – Lead Investigator Dr. Hans Ghayee
The absence of safe and effective treatment for metastatic pheochromocytoma/paraganglioma (PCC/PGL) that is especially prevalent in carriers of deleterious mutations in the subunit B of succinate dehydrogenase complex (SDHB) remains the highest clinical concern. With the support from the SDHB Pheo-Para Coalition, the research team from the University of Florida (UF) undertook a metabolomics approach to compare the metabolite contents of the tumors with and without SDHB mutation. Their analysis of the metabolome in cell lines and patient tumor samples with SDHB mutations demonstrated augmented activity of polyamine pathway upon SDHB deficiency.
In agreement with the metabolomic analysis, inhibitors of the polyamine pathway, N-alkylated polyamine analogues, proved to be remarkably potent against tumor cells with a loss of SDHB function, both in vitro and in the mouse xenograft model. The team is in the process of solving the mechanism on how this works. The goal is to see if compounds targeting the polyamine pathway can be brought to clinical trials in PCC/PGL safely.
Read More

Tufts Medical Center – Lead Investigator Dr. Arthur Tischler
Paragangliomas are rare neuroendocrine tumors that typically arise along the distribution of sympathetic nerves in the abdomen and chest and branches of cranial nerves in the head and neck. A paraganglioma that develops in the adrenal gland is called a pheochromocytoma. Paragangliomas can be caused by hereditary mutations of at least 19 different genes. Those caused by SDHB mutations are particularly aggressive and prone to metastasize. The development of effective treatments has been hampered by lack of valid models for pre-clinical testing and basic research.
The molecular and metabolic characteristics of SDHB-associated PCPG are fundamentally different from those of most other PCPG. Although researchers have attempted for many years to establish cell lines and xenografts from these tumors, until now there has been no model fully representing the distinctive characteristics of the actual tumors that occur in affected patients. Notably, attempts to establish mouse models have been unsuccessful. Using a novel strategy employing rats instead of mice, the Tischler laboratory recently established a cell line and xenograft model called RS0 (for rat Sdhb null), from heterozygous genetically engineered rats with a germline deletion in one Sdhb allele. Extensive, multiinstitutional, collaborative studies published in Endocrine Related Cancer in April 2020 showed that RS0 has lost the normal Sdhb allele, lacks Sdhb protein, and closely resembles SDHB-mutated human paragangliomas genetically and metabolically. It therefore appears to be the first example of a genetically and phenotypically valid model that many researchers in the field have been looking for. Fortuitously, a second model called RS1/2 developed in parallel from the same rat strain has lost the mutated SDHB gene and retains the normal one. RS1/2 may therefore serve as a control for experiments on RS0. Ongoing studies employ the RS0 and RS1/2 models to test prototype drugs using strategies that selectively target the distinctive vulnerabilities of Sdh deficient tumors and to determine the mechanisms of antitumoral effects by querying changes in relevant RNA and protein expression, and metabolic pathways critical to tumor growth. This integrated approach may lead to new strategies for slowing or preventing the growth of PCPG caused by mutations of the SDHB gene and improving the lives of affected patients.
Sdh-deficient paragangliomas are essentially a metabolic disease, characterized by expression of hypoxia-associated genes and altered metabolism that may result in a fragile, energy-compromised state. These characteristics suggest that drugs which compromise energy production or target hypoxic signaling pathways could be effective therapies alone or in combination. studies employing cultured RS0 cells have demonstrated antiproliferative and cytotoxic effects of several candidate drugs. Other laboratories in Australia, Europe and the United States have requested or already received the RS0 model for related studies..
The complete published paper describing the RS0 and RS1/2 models, which includes extensive background information, supplementary data and links to raw data files, can be accessed at https://doi.org/10.1530/ERC-19-0474.
Read More

Columbia University – Lead Investigator Dr. Tito Fojo
Our group is examining SDHB loss in pheochromocytoma in two projects. We have begun contacting patients and families to create a registry in which we will ask patients to contribute blood samples. From these samples, we will look for evidence of succinate excess in an effort to develop an earlier biomarker for the disease. We believe that several signature genes will be downregulated in cells exposed to chronic levels of succinate. In the second project, we have begun looking for drug candidates that could turn off the excess succinate. Such an agent could be treatment for patients with overt malignancy. However, a nontoxic, low dose agent without significant side effects could also be considered as a preventive measure. Our laboratory has screened a large number of compounds in collaboration with the National Center for Advancing Translational Sciences (NCATS, https://ncats.nih.gov/). Several hits emerged from this study and are being validated and pursued. Our overarching goal is to find new treatment options for SDHB-loss pheochromocytoma and to find an option that can be used very early in the natural history of the disease.
Read More
Stanford University – Lead Investigator Dr. Justin Annes
Annes Lab Research Abstract
Inherited mutations in Succinate Dehydrogenase genes (SDHx) are responsible for a constellation of tumors including pheochromocytoma and paragangliomas (PPGL), renal cell carcinoma (RCC) and gastrointestinal tumors (GIST). Notably, tumors that result from an inherited pathogenic SDHB gene mutation have substantial metastatic risk, estimated to be ~40-50%. Unfortunately, effective treatments for metastatic SDHB-related disease are lacking. Indeed, a critical step along the therapeutic development pathway is establishing an animal model that faithfully recapitulates disease and facilitates therapeutic discovery. Although, numerous efforts over the past 20 years failed to generate an SDHB-deficient PPGL mouse model, leading many to conclude its impossibility, we recently succeeded in producing the first SDHB-deficient PPGL mouse model by co-deletion of SDHB and the NF1 tumor suppressor gene. We found that isolated SDHB deficiency was not sufficient for tumorigenesis, despite sharing many key features of SDHB-deficient tumors, such as mitochondrial swelling, histone hypermethylation and succinate accumulation. These findings led us to conclude that SDHB-deficient tumorigenesis requires disruption of additional, unidentified, tumor suppressor gene(s) that promote cellular replication and enable succinate-dependent cellular and metabolic reprograming (DNA hypermethylation).
The Annes Lab is pursuing three key areas of investigation advance SDHB research and the care of SDHx familes.
Project 1. Develop enhanced mouse models of SDHB-deficient pheochromocytoma /paraganglioma (PPGL). Specifically, the Annes lab is testing the role of novel candidate tumor suppressor genes (TSGs) in tumor formation. We have selected candidate TSGs based upon suspicious findings which suggest they play a critical but unrecognized causative role in SDHB-PPGL formation, e,g. they demonstrate reduced expression in human SDHB-deficient PPGLs. These candidate TSGs are being tested individually and concurrently with disrupted SDHB expression in mouse chromaffin cells. This research objective is of critical importance to the SDHB field because advancing new depends upon an accurate understanding of how SDHB-PPGLs form and grow, and having an animal disease model in which new therapeutic candidates can be tested. Currently, the lack of an SDHB-PPGL animal model is slowing progress towards a cure.
Project 2. Perform chemical and genetic screening to identify drugs and drug-targets that are uniquely cytotoxic towards SDHB-deficient cells. We hypothesize that SDHB deficiency creates fixed, cell-autonomous vulnerabilities that are amenable to selective targeting, i.e. opportunities to realize synthetic lethality. We are exploiting the complementary strengths of chemical and genetic screening approaches to discover pathways that maintain SDHB-deficient cell viability. This research objective is critical to opening new avenues of therapeutic development for SDHB-cancers.
Project 3. Develop an experimental assay for determining the function (pathogenicity) of SDHB variants of uncertain significance (VUS) and assesses potential genotype-phenotype relationships. Because roughly forty percent of known SDHB mutations are VUSs, numerous at-risk patients/families are left in a quandary after receiving their genetic test results. They do not know whether they are at risk for tumors or not. We are developing an SDHB functional assay that can be used to determine the pathogenicity of VUSs and improve care for currently unrecognized, but at-risk, SDHB families. Furthermore, our experimental system provides an experimental platform for uncovering potential genotype-phenotype relationships.
Collectively, the Annes Lab is addressing high priority fundamental, translational and clinical gaps in our understanding of SDHB-cancers with the intent of transforming the future outlook for affected individuals.
Read More